Informed Design of Bioanalytical PCR Assay Testing Parameters
Explore validation parameters for PCR-based PK assays to best mitigate regulatory risks.

Cell and gene therapies (CGTs) are one of the fastest growing areas in molecular research and human therapeutics. These treatments use a vector to introduce the desired DNA, or nucleic acid code, to replace or modify protein expression or use cells to alter/restore a specific cell type. Since chimeric antigen receptor T cell (CAR-T) therapy was first approved in 2017, there has been a marked increase of cell and gene therapy studies resulting in significant changes in the way diseases are treated as well as patient outcomes.
Polymerase chain reaction (PCR)-based bioanalytical testing is the cornerstone of evaluating delivery, safety and efficacy of CGTs; however, specific regulatory guidance for analytic methods the like PCR assay has yet to be established. There are over 2,000 CGTs currently being evaluated in clinical trials, with more than 200 in Phase III and 10-20 per year estimated to come to market by 2025.
PCR testing: The cornerstone tests for evaluating CGTs
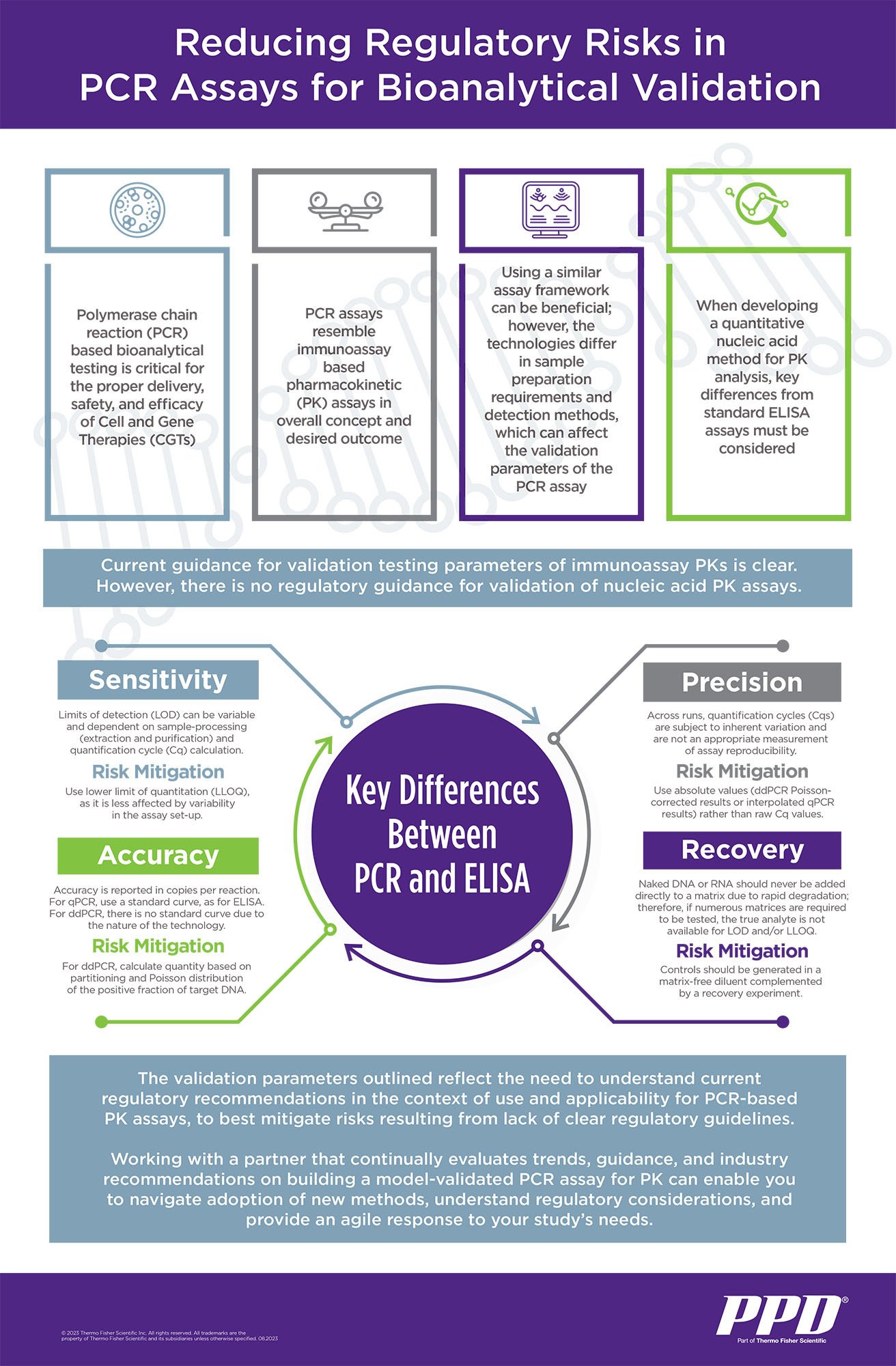
At several steps during CGT development and testing, sensitive, accurate and precise quantitation of CGT drug vectors is required. However, given the lack of consistent and specific regulatory guidance for PCR-based bioanalytical methods, regulatory guidance available for other quantitative assays must be referenced when planning analytic methods for these studies. Parallels between pharmacokinetic (PK) analyses for CGTs and immunotherapies, conducted through a PCR assay and a plate-bound antibody assay, respectively, can inform a successful approach. However, key differences between a PCR assay and an immunotherapeutic assay must be considered. It is critical to understand guidelines for designing these studies and the key parameters for successful quantitative nucleic acid analysis method development.
Addressing crucial differences between a PCR assay and a standard ELISA assay with respect to sensitivity, accuracy, precision and recovery will help build a strong case when submitting data and results to the U.S. Food and Drug Administration (FDA). Without such forethought, extensive future redesign and/or repetition of assays may be required. In addition, laboratories should be positioned to have an agile response to rapidly adopt any new methods once regulatory guidance is published. Working with experienced providers can help clients build data sets that satisfy current and future FDA requirements. It is this kind of familiarity and knowledge of FDA filings that an experienced provider can offer their clients to save both time and money, while avoiding bottlenecks with future filings.
Emerging areas of pharmaceutical research not only bring novel opportunities but also new regulatory guidance. The risk remains that once clear guidance is set, assays, including the PCR assay, validated to current practice may no longer be sufficient. In this uncharted landscape, it is critical that clinical trial teams possess familiarity with past regulatory guidance as well as new technologies and methods for reducing regulatory risks.
Want to stay abreast of new methods, regulatory considerations and ways to provide an agile response to your study’s needs?
Learn more about how PPD™ Laboratory services can enhance your CGT projects.