
Implementing the Estimands Framework in Clinical Trials

In this blog post, our cross-functional estimands working group discusses the benefits an estimand framework can bring.
Our working group has developed case study examples and training materials. It has also streamlined our protocol development process to implement the estimands framework in any clinical setting and to support sponsors in their protocol development.
We have developed a tool to aid in-depth thinking and discussions between clinical and statistical experts and updated our templates to clearly document estimands aligned to clinical trial objectives.
What is an estimand?
Simply put, an estimand clarifies what we want to find out about efficacy, safety or pharmacokinetics in clinical study protocols. Regulators are now requiring study objectives and endpoints be expanded to detail preplanned estimand(s) at the protocol writing stage. Clinical studies aim to quantify the benefit, or effect, of the investigational product. Clarifying these questions in the protocol helps to precisely define the parameter to be estimated, which is called the estimand. According to regulatory guidance1, an estimand should be defined precisely, including five attributes (A to E below):
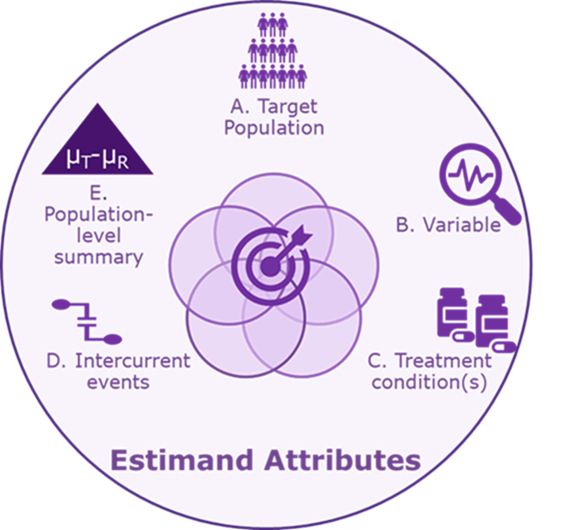
A. Target population: The full population of all patients who would meet the study’s eligibility criteria.
B. Variable: The endpoint measured for the individual patient.
C. Treatment condition(s): The investigational and reference products. These may be administered in addition to standard care or rescue medication.
D. Intercurrent event(s): This is defined in ICH E9(R1) as “events occurring after treatment initiation that affect either the interpretation or the existence of the measurements associated with the clinical question of interest.”
Examples of intercurrent events include treatment discontinuation due to adverse events or lack of efficacy, rescue medications and death. This attribute is not merely listing the intercurrent events that are expected to occur in a given clinical setting but also specifying how to account for each intercurrent event in relation to the given estimate.
There are five potential strategies to address intercurrent events: treatment policy, hypothetical, composite, principal stratum and whilst on treatment. Each intercurrent event may require a different strategy.
E. Population-level summary: The summary of the variable in the target population which allows comparison between treatment conditions. Population-level summaries may include a difference in means, response rate and difference in median survival times.
Many experienced clinical trial designers are not yet familiar with attribute D: how to account for intercurrent events in the scientific question of interest. Each of the attributes are explained further through a COVID-19 vaccine example below.
Case study: COVID-19 vaccine clinical study that uses estimands
The primary objective of this example Phase 2 COVID-19 vaccine study is to compare immunogenicity of two alternative test vaccines which may differ in terms of vaccine dose levels, adjuvant choice and antigen components. The protocol is designed to compare these test vaccines (New-CoV-Vac-1 and New-CoV-Vac-2) with each other and with placebo as assessed by SARS-CoV-2 neutralizing antibodies at two months after the first vaccination.
Example attributes of the estimand
A. Target population: Healthy adults at low risk of SARS-CoV-2 infection, excluding those with active or prior infection at the time of the first vaccination.
B. Variable: Neutralizing antibody titers to SARS-CoV-2 at two months after first vaccination.
C. Treatment conditions: New-CoV-Vac-1, New-CoV-Vac-2 or placebo with vaccinations on day 0 and homologous booster on day 21.
D. Intercurrent events: The following intercurrent events could impact the antibody levels achieved:
- Missing the second vaccination
- Receiving of immune-modifying drugs or vaccines
- Subsequent infection with SARS-CoV-2
Strategies for each intercurrent event should also be considered as part of this attribute. Here, interest lies in estimating the “hypothetical” effect of the vaccines on SARS-CoV-2 neutralizing antibodies, assuming both day 0 and day 21 vaccinations were received without subsequent SARS-Cov2 infection and without any influence from any other immune-modifying drugs or vaccines. Statisticians should note that the “hypothetical” estimand can be estimated using a repeated measures model or multiple imputation methods excluding data points after relevant intercurrent events.
E. Population-level summary: Ratio of geometric means of the neutralizing antibody titers (New-CoV-Vac-1/placebo, New-CoV-Vac-2/placebo and New-CoV-Vac-1/New-CoV-Vac-2).
How are estimands documented in the protocol?
We recommend documenting the estimands associated with each primary and key secondary objective in a protocol table to highlight the alignment between each objective and the related estimand(s):
| Objective | Estimand description (including endpoint) |
|---|---|
| Immunogenicity: To compare the New-CoV-Vac-1 and New-CoV-Vac-2 vaccinations with each other and with placebo, as assessed by neutralizing antibodies against SARS-CoV-2 in healthy adults at low risk of infection, two months after first vaccination. | Immunogenicity Estimand: Ratio (New-CoV-Vac-1/placebo, New-CoV-Vac-2/placebo and New-CoV-Vac-1/New-CoV-Vac-2) of geometric means of neutralizing antibody against SARS-CoV-2 infection at two months after first vaccination in healthy adults at low risk of infection. The hypothetical strategy is used to estimate antibody levels if both scheduled vaccinations are received (on day 0 and day 21) and without any influence from immune-modifying drugs or subsequent infection. |
Similarly, the protocol statistical methods should include details of the main estimation and sensitivity analyses aligned to each estimand.
What is the regulatory position in relation to estimands?
A final version of ICH E9(R1) was adopted by ICH on Nov. 20, 2019. In 2020, the U.S. Food and Drug Administration (FDA) and other regulators around the globe are in the process of implementing the estimands framework; for example, the European Medicines Agency (EMA) set July 30, 2020, as their “legal effective date,” and will be expecting estimands documented in new protocols.
Summary of benefits
The documentation of estimands in protocols is expected to grow rapidly as more sponsors see the benefits that the estimand framework can bring. These benefits include adding clarity to study objectives, informing study design and data collection practices for critical data, and driving choice of statistical methods. This brings transparency in reporting meaningful treatment effects which inform clinical decision-making and ultimately smoother regulatory approvals. The PPD™ clinical research business of Thermo Fisher Scientific encourages early discussions around estimands and has now implemented estimands in protocol synopses and protocols across multiple therapeutic areas.