
New European Union Regulation to Change How Clinical Trials are Conducted

New clinical trial regulations are coming to the European Union in 2022. In this blog, our experts explain what is changing and how we can help you prepare for and navigate the transition.
Authors:
- Angela Nelson, executive director, regulatory affairs
- Rina Kacha, senior manager, regulatory affairs
- Tijana Pajic, senior manager, regulatory affairs
In an effort to increase transparency and ensure the highest standards for patient safety, the European Union (EU) will undergo a major change in the way clinical trials are conducted. The existing EU Clinical Trials Directive (2001/20/EC) shall be repealed, and the EU Clinical Trials Regulation (EU CTR) (Regulation EU 536/2014) will become applicable as of Jan. 31, 2022.
The process to harmonize the assessment and supervision of clinical trials will be challenging, yet the results — for example, improved collaboration, information sharing and decision making — will be well worth the effort.
To help clinical trial sponsors prepare for and successfully transition through these upcoming changes, our experts are sharing guidance on the major changes, including:
- The process for submission of clinical trial applications in the EU
- The introduction of new classifications of trial types, such as low interventional trials
- The introduction of the lay language
- The use of the EU clinical trial information system (CTIS) for centralized submissions
Q: How will timelines for response to queries be impacted?
A: The EU CTR provides clear timelines for the evaluation of clinical trial applications (CTA). If there is a request for information, there will be a maximum of 12 days to respond. A failure in response will lead to automatic withdrawal of the application in all member states.
In short, this means that revisions, translations and redactions need to be completed within the 12 days allowed to provide the response submission.
Q: What’s the future of the voluntary harmonization process (VHP)?
A: The last day for acceptance of VHP submissions was Oct. 15, 2021. Trials submitted via the VHP will qualify for transition from the CTIS implementation date of Jan. 31, 2022. In the meantime, until VHP trials are transitioned, any substantial amendments submitted to VHP trials will be assessed in accordance with the clinical trial directive by individual member states.
Q: How have substantial modifications been affected?
A: The EU CTR allows less flexibility when it comes to submitting substantial modifications. In fact, substantial modifications can only be submitted if all member states have issued a decision on a previous assessment and at least one of those decisions is positive. This means that it could take up to 94 days before the next substantial modification can be submitted. Therefore, it is recommended that modifications be planned in advance to avoid delays.
Early development of strategies which accommodate substantial modifications across the lifecycle of multiple clinical trials for an asset within a portfolio of trials is critical to minimize risk. A cascade of increased risk can occur due to a single substantial modification failure across the entire portfolio if not adequately addressed.
To avoid these hurdles, sponsors should consider partnering with a CRO that can support the development of an appropriate regulatory strategy for the scientific content found in part one (e.g., protocol, IB, IMPD) and/or the national content found in part two (and/or part two of the application dossier, e.g., patient information, insurance, trial site, investigator information).
Q: What are the public disclosure requirements?
A: The EU CTR promotes transparency of clinical trial data, allowing public access to information — except for sensitive and commercially confidential data (such as the quality IMPD) — via the CTIS. During the clinical trial application, sponsors have the option to provide documents with confidential information redacted in addition to the complete document for review. Sponsors will also have the option to submit a justification requesting the publication to be deferred for up to seven years depending on the trial type.
Q: What are the plain language requirements?
A: To increase access and transparency, the EU CTR promotes the requirement for use of plain language for some documents. In fact, the regulation requires that the protocol synopsis and summary of the clinical trial results be understandable for laypersons. The documents should be provided in the local language of each EU country in the trial and submitted through the CTIS.
Q: How is the labelling of clinical trial supplies affected?
A: When transitioning a clinical study to the new EU CTR or submitting an initial application to the EU CTR, updates are required to the label format for investigational medicinal products and auxiliary medicinal products to include “period of use” (expiration or retest date).
Q: Is there a transition period for new applications?
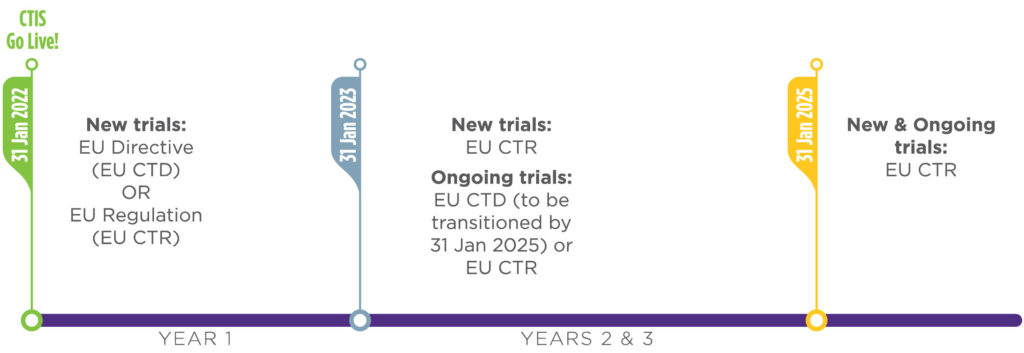
A: All eligible clinical trials will be conducted under the EU CTR by Jan. 31, 2025. The European Medicines Agency has provided a phased implementation for the regulation in which during the first year of the implementation, from Jan. 31, 2022, until Jan. 31, 2023, sponsors may elect to submit applications for new clinical trials under the current EU Clinical Trials Directive (EU CTD) or the new EU CTR.
Also, during this first year sponsors may elect to transfer eligible existing clinical trials to the EU CTR if they wish to do so. However, after the first year of implementation (after Jan. 31, 2023), all new applications for clinical trials must originate under the EU CTR.
Additionally, clinical trials in conduct under the EU CTD after Jan. 31, 2023, will be required to transition to the EU CTR prior to addition of a new member state(s). All remaining eligible trials — with durations extending past Jan. 31, 2025, that have not transitioned as part of the first phase — will be required to transition their authorization and remaining conduct from the EU CTD to the EU CTR no later than Jan. 31, 2025.
Q: What about auxiliary medicinal products (AMP)?
A: An AMP is described in the protocol as used for background treatment such as challenging agents, rescue medications or tools to assess clinical trial endpoints. Non-authorized AMPs will be required to be labeled per annex VI, certified by the qualified person to ensure compliance with good manufacturing practice, traced and reported on in case of any adverse events.
Q: How will safety reporting change?
A: The new regulation aims to streamline safety reporting to prevent companies from having to make separate safety-related submissions per study with the same investigational medicinal product (IMP). A new EudraVigilance database will be implemented to submit serious adverse events and suspected unexpected serious adverse reactions. For clinical trials that involve more than one IMP, a single safety report can be submitted via the EudraVigilance system. Annual safety reports, including the development safety update report, will be submitted annually through the CTIS.
Q: What about the United Kingdom (U.K.)?
A: The Medicines and Healthcare Products Regulatory Agency in the U.K. has been heavily involved in the preparation of the EU CTR. Due to Brexit, the new regulation will not apply in the U.K. However, the Combined Ways of Working Pilot (CWOW) has been introduced to prepare for the change, resulting in a single U.K. decision (combined regulatory and ethics) on a clinical trial. As of Jan. 1, 2022, all new CTAs must be submitted via the CWOW process.
Q: What other elements need to be considered?
A: A low interventional trial is a new definition under the EU CTR that includes trials with IMPs (excluding placebos) that are authorized, are used in accordance with the terms of the marketing authorization or are evidence-based and supported by published scientific literature. Interventional and low-interventional trials are in scope of the EU CTR, while non-interventional trials will continue to follow national legislation. Protocols should be reviewed to determine the definition of the applicable clinical trial.
Q: How can sponsors successfully manage the EU CTR change?
A: The consequences of sponsors not adequately preparing are many: delays, increased costs and, ultimately, delayed therapeutic development. Forward-thinking companies and CROs have been preparing for these changes as the EU CTR takes full effect. In the coming year, the PPD™ clinical research business of Thermo Fisher Scientific will advise sponsors on the transition of their trials, provide the necessary support for ongoing and new trials as part of clinical development plans, and assist with trials that need to transition over the three-year period.