
Success Factors for eCTD Implementation in Mainland China and Taiwan

The electronic common technical document (eCTD) is widely accepted and implemented by regulatory authorities in many countries, and its application in mainland China and Taiwan has been long anticipated. Jeffery Xu, publishing manager, and Rebecca Sagosz, regulatory publishing project consultant, discuss advances in eCTD implementation in mainland China and Taiwan and detail three critical success factors for pharmaceutical companies’ readiness.
What is eCTD?
eCTD is a standard format for submitting regulatory dossier (such as applications, amendments, supplements, reports, etc.) to regulatory authorities. The benefits of eCTD are well established; for example, it supports global application, improves efficiency of data exchange between pharmaceutical companies and regulatory authorities, and enables quicker reviews, thus shortening time to approval.
In mainland China, since Dec. 29, 2021, the National Medical Products Administration (NMPA) has accepted eCTD marketing authorization applications. Acceptance of eCTD clinical trial applications is expected later this year.
The Taiwan Food and Drug Administration (TFDA) expects to implement eCTD some time in 2022 (specific date yet to be announced), initially for new drug application submissions (starting with new chemical entities).
Here is what pharmaceutical companies need to know about the eCTD rollout in China and Taiwan
Mainland China
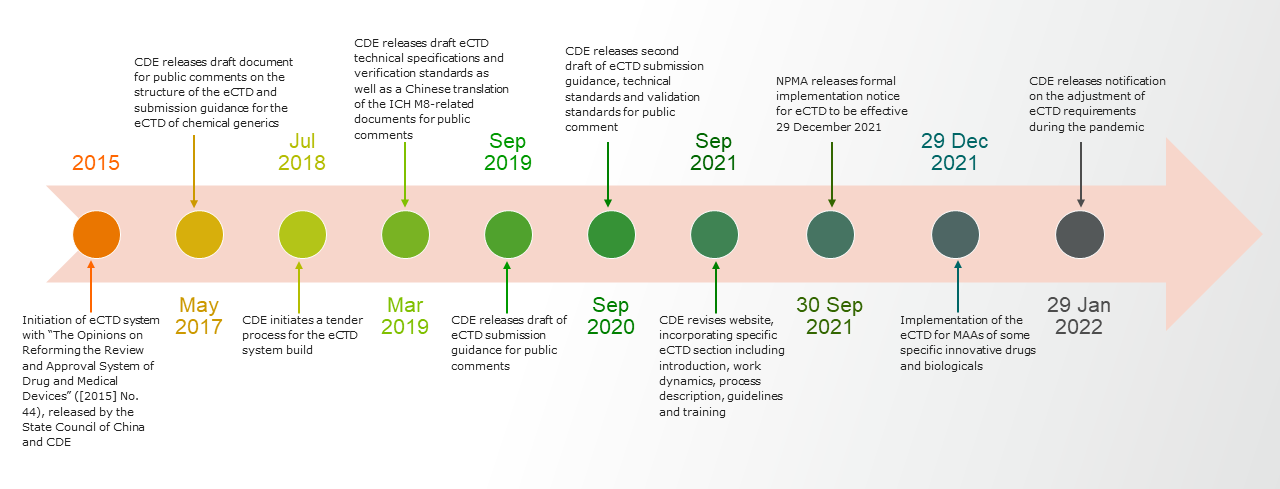
Efforts over many years have led to the long-awaited eCTD implementation by the NMPA (overview in the figure below). As of Dec. 29, 2021, the NMPA has implemented use of the eCTD for marketing authorization applications of some specific innovative drugs and biologicals.1 Until mainland China implements the use of eCTD for clinical trial applications, expected sometime in 2022, the NMPA will require applicants to submit both a hard copy and eCTD format application.
Based on the NMPA technical specifications, published September 2021, NMPA’s criteria are more aligned to the U.S. Food and Drug Administration (FDA) eCTD specifications rather than those of the European Medicines Agency (EMA).1 However, there are significant differences in NMPA submission requirements compared to both the FDA and the EMA, including:
- The NMPA requests that all documents be translated into Chinese with original documents in English. Other languages can be attached for reference. An electronic file naming convention exists to distinguish the language of a given document, with Chinese versions labeled as “zh” and English versions labeled with ”en.” Documents in languages other than Chinese will not be subject to certain eCTD validation standards.
- The eCTD is submitted through physical electronic media (e.g., CD-R, DVD+R and DVD-R), with hard copy materials to be submitted within five business days of the acceptance of an eCTD submission.
- Both the EMA and the FDA utilize “gateway” platforms for secure electronic submissions; a gateway submission is not currently an option in China, but development of an electronic submission system is being prioritized by the China Center for Drug Evaluation (CDE).
- Other significant points of difference include the file structure, study tagging file, extension node and clinical trial basis.1
On Jan. 28, 2022, the CDE issued further guidance on specifications to apply during the pandemic only. This guidance covers details such as file format, PDF version, file name and path, file size, bookmarks and hyperlinks, page number compilation, file compression and encryption, CD requirements, virus check and more. The requirements are vast and wide-ranging, but a big step in paper submission, further signaling China’s firm intention to approaching actual implementation of eCTD.
Taiwan
In Taiwan, the implementation of eCTD is under the jurisdiction of the TFDA. While progress has been made, it has not matched that of NMPA, and key details remain to be finalized.
In December 2020, the TFDA issued eCTD specifications and validation criteria as a step toward adoption of the eCTD. This included “Guidelines for Electronic General Technical Documents for Drug Inspection or Registration,” which contained recommendations and principles for submission documents for new biological and generic drug registrations, as well as post-marketing changes. Note that this guidance is not yet final, and as a result, e-readiness is challenging. It defined modules 2 to 5 in accordance with ICH eCTD specification v3.2.2, as well as a module 1 that complies with Taiwan’s regional regulatory requirements for establishing submission data. In addition, although the TFDA has indicated its intention to implement the eCTD with all types of CTA submissions, the timing for this remains to be defined.
Similar to the NMPA, TFDA’s criteria are more aligned to the FDA’s eCTD specifications than the EMA’s eCTD process. In contrast to NMPA processes, the TFDA has an electronic submission platform, “ExPress,” for life cycle management and transmission of information. The TFDA can accept both Chinese and English languages; however, the application form, cover letter and any patient-facing documents must be in Chinese.
Are you ready?
While eCTD format improves efficiency of filing and review, it also brings challenges. For example, applicants must develop an appropriate eCTD submission strategy and end-to-end submission processes, including document preparation and quality control processes.
Here are three critical e-readiness success factors for pharmaceutical sponsors and regulatory agencies:
- Experienced personnel: Sponsors and agencies should have the most efficient and knowledgeable staff to provide on-time deliverables with premium output. Engage personnel with professional electronic publishing and submission skills, and clarify job responsibilities, such as eCTD data review, verification, submission or archiving. Continued training of personnel, such as regulatory training and participation in academic forums, will enable them to keep current with global electronic publishing and submission guidelines and specifications.
- Workflow: Create internal standard operating procedures, guidance documents and/or job aids — including for templates, document review lists, document naming convention, document granularity, as well as appropriate document flow and review processes. Appropriate project management will enable timely delivery and high-quality output.
- Platform and system: Implement appropriate eCTD software as well as related document processing, electronic gateway document storage and archiving in a document management system and other tools to meet e-readiness goals. Always ensure software is current, enabling compliance with the latest electronic specifications and validation criteria.
The assistance of professional third-party outsourcing services to provide accurate and efficient eCTD services will be essential for pharmaceutical companies to assure timely approvals, especially as many organizations are still building internal expertise, and information continues to emerge.
The PPD™ clinical research business of Thermo Fisher Scientific remains encouraged by the determination and action of NMPA and TFDA to align with international standards and promote innovation in this area. Our customers remain ready to move forward, as our experts proactively keep abreast of industry trends, evaluate the latest policy requirements and adapt processes to support eCTD submissions in mainland China and Taiwan.
Seeking more regulatory expertise? Check out our latest regulatory insights.