
The Drug Discovery and Development Process
The challenges and costs behind bringing new medicines to market
The drug discovery and development process is long, challenging and rewarding at the same time. New medicines save lives and improve the quality of life for millions of people. The development of prescription and over-the-counter drugs, vaccines, cell therapies, medical devices and surgical or other treatment procedures can stop diseases from getting worse or even reverse their course.


Drug discovery
In the discovery stage – the first step in bringing a new drug to market – researchers evaluate compounds to determine which could be candidates for development as medical treatments.
The process begins with the identification of a new target molecule, a protein or other molecule involved in the disease process. Once a target molecule is identified, scientists must design and synthesize a new compound that will interact with the target molecule and influence or inhibit its function.
Researchers use several methods in the discovery process to identify and evaluate potential treatments. They include:
- Testing many molecular compounds to find if they offer possible benefits against diseases
- Re-testing existing treatments to see if they offer benefits against other diseases
- Using new information about diseases to design products that could stop or reverse the effects of the disease
- Adopting new technologies to treat diseases, such as those that provide the ability to target specific sites within the body or manipulate genetic material
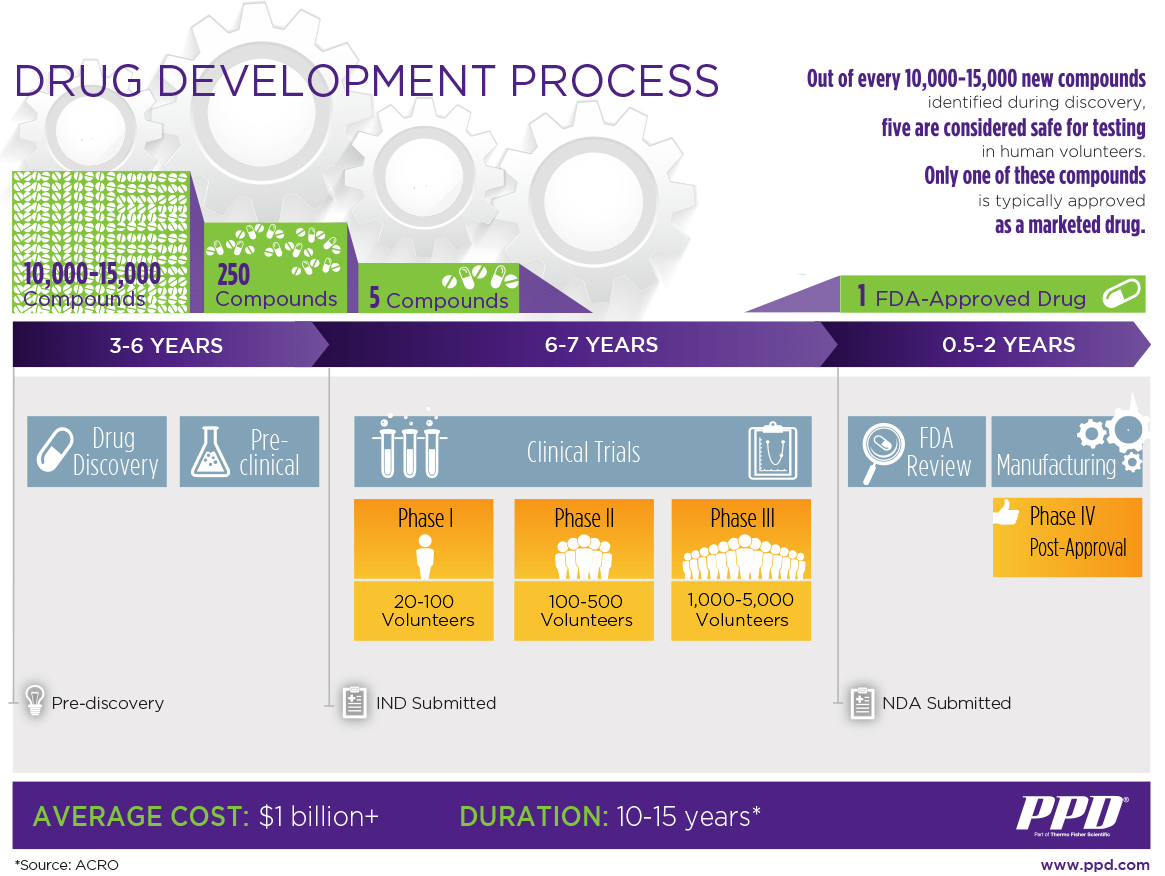
During the discovery stage, researchers may look at thousands of compounds to identify candidates for development as therapeutic treatments. Typically, only a few compounds look promising and call for further study. The Pharmaceutical Journal, a research publication based in the United Kingdom, estimates that for every 10,000 compounds tested in the discovery stage, only 10-20 compounds move on to the development phase. And only about half of those entering the development phase ultimately proceed into preclinical trials.


Preclinical research development stage
The next step is to conduct non-clinical studies to assess the toxicity and activity of the new compound in animals (referred to as in vivo testing) and human cells (housed in glass or plastic laboratory vessels, which is known as in vitro testing). Animal models that mimic human conditions, such as knockouts or genetically modified mice, may be used during this stage. The PPD™ clinical research business of Thermo Fisher Scientific does not conduct trials in animals.
Toxicity studies typically involve giving the new compound to animals at increasing doses to see how they respond. Activity or pharmacological effect studies are conducted to assess the ability of the new compound to treat the target disease in animals (seeing if it improves their condition). These studies can take several years to complete.
Study of the pharmacology and toxicology of the lead compounds
The results of preclinical studies must provide detailed information on the drug’s underlying pharmacology and toxicity levels. To ensure the consistency of results, regulatory agencies throughout the world — including the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA) and other national agencies — require that researchers use good laboratory practices (GLP) for preclinical laboratory studies. The GLP regulations set minimum basic requirements for:
- Study conduct
- Personnel involved
- Facilities
- Equipment
- Written protocols
- Operating procedure
- Reports
GLP standards also specify a system of quality assurance and oversight for each study to ensure the safety of regulated products.
If the new compound is found to be safe and effective in human cells, all findings of the preclinical research are submitted to regulatory authorities for permission to conduct clinical testing in humans.

Development of the dosage formulation
Proper dosing determines medication effectiveness. Once researchers identify a promising compound for development, they conduct experiments to gather information on:
- How it is absorbed, distributed, metabolized and excreted
- Its potential benefits and mechanisms of action
- The best way to administer the drug (such as by mouth or injection)
- Side effects or adverse events (toxicity)
- How it affects different groups of people (such as by gender, race or ethnicity) differently
- How it interacts with other drugs and treatments
- Its effectiveness as compared with similar drugs
Manufacturing of the drug according to GMP standards
The drug developer also is expected to provide data on the production method and data generated from testing the compound as it is developed. It is initially made in small batches at the benchside. Then during the manufacturing and development process, larger batches are generated. This scale-up of a drug must be done using Good Manufacturing Practices (GMP). Consideration should be given to future manufacturing suitability, commercial viability and cost-effectiveness. Regulatory approval is for the final product and the utmost care must be used in producing and testing the product to ensure that each new batch meets the same standards of purity and quality.

Application to regulatory authorities
Individual countries have their own regulatory authorities for drug development. Before a trial initiates, the drug developer weighs the foreseeable risks against the anticipated benefits for the individuals participating in the trial and for society at large. The risks must outweigh the benefits for the trial to proceed.
In the U.S., the application for clinical testing in humans is called the Investigational New Drug (IND) application, which is submitted to the FDA. In the IND application, developers must include:
- Animal pharmacology and toxicity data
- Data from any prior human research
- Clinical protocols (study plans) for proposed human studies to be conducted
- Information about the principal investigator(s)
- Manufacturing information
The FDA then conducts an extensive review of the IND, and, after 30 days, can respond in one of several ways:
- A temporary hold until additional information is obtained, or a stop of the investigation entirely
- Approval of the IND and commencement of clinical trials


Clinical drug development process
Clinical trials refer to studies with human participants that answer specific research questions related to a medical product. These trials follow a study plan, called a protocol, that is developed by the researcher or manufacturer to describe exactly how the clinical trial will be conducted. It details key study objectives, design and statistical considerations to ensure the safety of participants and the integrity of the data collected during the study.
Before a clinical trial begins, researchers (led by a principal investigator, who is usually a physician) review prior information about the drug to develop research questions and objectives. Then they decide:
- Who qualifies to participate (selection criteria)
- How many people will be part of the study
- How long the study will last
- Whether there will be a control group (the group of participants that receives standard treatment or a placebo) and other ways to limit research bias
- How the drug will be given to patients and at what dosage
- What assessments will be conducted, when, and what data will be collected
- How the data will be reviewed, analyzed and reported
Clinical trials occur in three phases. Speed and efficiency during this process are critical for the commercial success of new treatments and therapies. One reason is that the time spent in the development process counts against the drug’s patent protection period after it goes to market. When the patent expires, generic competition can sharply reduce sales revenue.


FDA drug application approval process
If the new compound is successful in several large, controlled clinical trials, the collected data are then combined with preclinical and other supportive real-world evidence to file a Marketing Authorization Submission seeking approval for public use of the new medicine.
Regulatory authorities evaluate all of the available data for safety and efficacy prior to approval. In the U.S., this requires submission of a New Drug Application (NDA) or Biologics License Application (BLA). The latter must be submitted for therapeutic biological products including (but not limited to) monoclonal antibodies, cytokines, growth factors, enzymes, immunomodulators, proteins and non-vaccine therapeutic immunotherapies.
The drug developers must include reports on all clinical results, data and analyses, as well as:
- Proposed labeling
- Safety updates
- Drug abuse information
- Patent information
- Any data from studies conducted outside the U.S.
- Institutional Review Board compliance information
- Directions for use
- Potential interactions with other medications
The FDA’s regulatory approval process for new drugs is rigorous, and the application is categorized as standard, fast track, breakthrough, accelerated approval or priority review depending on the drug’s relevance and necessity for patients. If standard or priority review is required, the approval timeline may take a year or more. Fast track, breakthrough, or accelerated approvals may occur sooner. (The FDA can also issue an emergency use authorization of an investigational drug in an urgent situation, such as during the COVID-19 pandemic, without the obligation to submit an NDA.)
If the FDA approves the new compound, the drug developer is clear to market it for patients. Upon approval, manufacturing operations can scale up from clinical trial size batches to commercial size batches. The larger manufacturing process and all of its components are validated to ensure results are consistently in line with the clinical trial batches with regard to the drug’s purity and quality. Other considerations include:
- Printing of final product label information, packaging and artwork
- Product storage, shipping and distribution arrangements
- Production staff and quality team availability
Post-approval monitoring and research
The true picture of a product’s safety evolves over time as more patients use the product and more post-marketing safety data are collected. Post-approval Phase IV monitoring aims to understand additional information about the product over the long term, including the drug’s safety, effectiveness and overall balance of benefits and risks in expanded patient populations and in real-world clinical use. The FDA requires drug companies to monitor the safety of new drugs using the FDA Adverse Event Reporting System (FAERS) database.
Examples of data obtained from this phase may be unpredicted serious side effects, interactions with other drugs, modifications to dosage and potential alternate uses (which would require the filing of a new IND). If a significant problem arises, the FDA could add caution statements to the drug dosage or usage information or take other measures. And if a developer wants to make changes in formulation, labeling or dosage strength, it must file a supplemental application and have the change approved by the FDA.


Q&A
What is the difference between drug discovery and drug development?
Discovery is the first step in bringing new treatments and therapies to market. In this stage, researchers evaluate thousands of compounds to determine which could be candidates for development as medical treatments. Often, only a few compounds look promising and call for further study.
Once a promising compound is identified, it moves into the drug development phase, which includes all the research steps that must be completed to transform the compound from a therapy candidate to a product approved for marketing by the appropriate regulatory authorities.
Why is drug discovery hard?
Many thousands of new chemical compounds are created and tested to identify those that have potential. But fewer than 0.001% of all compounds made ever progress to testing in humans. A new drug that shows potential will be put through a battery of laboratory and animal tests before being given a clinical trials authorization.
Why does drug discovery and development take so long?
In addition to the many tests conducted during discovery, every phase of the development process requires overarching consideration for the safety of participants and the integrity of the data collected during the research study. Each step must be carefully monitored for adverse events before proceeding to the next phase; there can be no shortcuts.
What is the probability of success in drug discovery?
During drug discovery, thousands of compounds may be potential candidates for development as a medical treatment. After early testing, however, only a small number of compounds may look promising and call for further study. It is estimated that for every 10,000 compounds tested in the discovery stage, only 10-20 compounds move on to the development phase.
Why is drug development so expensive?
There are many costs and implementation challenges associated with bringing a new drug to market. The main reasons for high costs are the need for multiple clinical trials in order to provide quality data in humans at large scale, and the cost of manufacturing the product in large batches for clinical use (Phase III and post-marketing).
A great deal of money is invested to evaluate promising compounds that ultimately do not make it to market. In Phase I clinical trials, approximately 70% of drugs move to the next phase. But in Phase II, only about 33% of drugs move to the next phase, and only about 25-30% of Phase III drugs move on to the approval process, where they still can be rejected. Thus, the likelihood of a drug making it all the way to Phase III clinical trials is just 12%.1
Cost control is a significant concern, because if the compound does not successfully make it through the development process and approval stages, the process must begin again on another compound, and the time, money and resources invested may be lost. However, many drug developers learn a great deal through failures, and in many cases, the lessons they learn drive subsequent breakthroughs and innovations.



