
European Union Clinical Trials Regulation: Clinical Trials Information System

In our ongoing series on the European Union Clinical Trials Regulation (EU CTR), experts focus on the key features of the Clinical Trial Information System (CTIS) and provide recommendations to sponsors for overcoming challenges.
Authors:
- Rina Kacha, senior manager of regulatory affairs
- Tijana Pajic, senior manager of regulatory affairs
- Marko Drobec, senior manager of process & system optimization
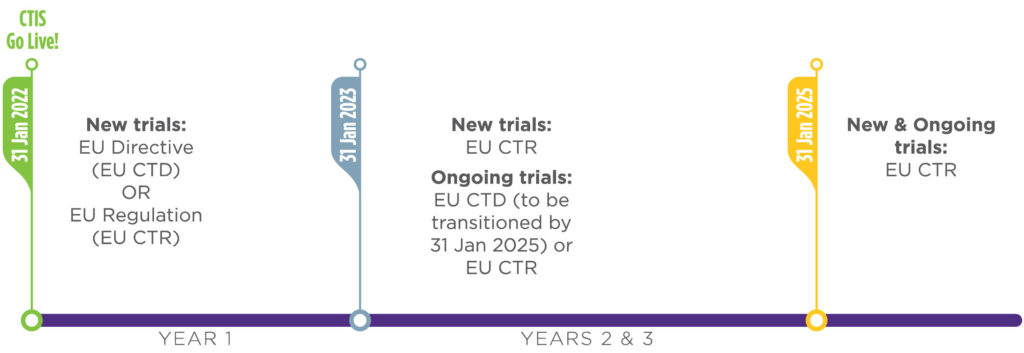
The application of the European Union Clinical Trials Regulation (EU CTR) and Clinical Trials Information System (CTIS) on Jan. 31, 2022, will result in considerable changes to the way clinical trials are conducted in the EU.
Specifically, the CTIS will serve as a single entry point for clinical trial information in the EU and the European Economic Area (EEA).
By utilizing an electronic submission platform for clinical trial applications and housing all modifications to regulatory authorities and ethics committees in EU/EEA countries, the CTIS will fully enable electronic exchange of information between sponsors and member states, facilitate increased transparency of clinical trials and provide a harmonized safety assessment to enhance patient safety.
While the benefits of the CTIS implementation are numerous, the new platform can be difficult to navigate and will require sponsors to make substantial adjustments.
To help with this transition, our experts are providing guidance on the use of the CTIS workspace and approaches to user management:
CTIS user roles
To access a CTIS workspace, an active European Medicines Agency (EMA) account is required. The CTIS has 49 roles across the sponsor and authority workspaces, including administrator and business roles. Currently up to 18 sponsor roles are foreseen in CTIS, with roles and access rights varying based on the activity the user will perform (viewing, preparing or submitting information).3 The sponsor administrative role is a high-level role with the authority to assign all other medium-level administrator and business roles, including preparer, submitter and viewer.4
CTIS user management
To meet the needs of the different types of sponsor organizations that will use CTIS, two user management approaches exist from which sponsors can select: i) organization-centric and ii) trial-centric.1 EMA recommends adopting the organization-centric approach.3
However, before using the CTIS, sponsors should carefully consider the advantages and disadvantages of each approach as outlined below:
Organization-centric vs. trial-centric approaches at a glance2,3
| Organization-Centric | Trial-Centric |
| Suitable for large commercial sponsor organizations managing many users and trials | Suitable for non-commercial sponsors managing a small number of users and clinical trials |
| Optional for sponsors, but mandatory for marketing authorization holders, member states and ethics committees | Available only for sponsors |
| Users will need to have roles assigned by the high-level administrators or delegated medium-level administrators to perform any action in the system | The default approach if the organization for which a clinical trial application is created does not have a high-level administrator |
| The high-level administrator will be able to manage and monitor all users within their organization | Follows a bottom-up model, since a high-level administrator is not required, which increases the agility when submitting a clinical trial application or when managing a small number of users5,6 |
| Administrator users can assign a role to users for all or specific trials that the organization manages5,6 | The management of users is done by the clinical trial administrator role, which is automatically created at the trial level |
Organization-centric approach vs. trial-centric approaches advantages and disadvantages2,3
| Organization-Centric Advantages | Trial-Centric Advantages |
| • Provides the opportunity to manage access and roles across trials • Provides organization/member states user and workload visibility • Improves security, as access to trials is centrally managed and enabled by role assignment/approval • In the sponsor user group, it ensures less duplication of data and data quality by having a high-level administrator, preventing the submission of clinical trial applications using any sponsor organization | • Faster and less complex initial application submission process, as no validation process is required |
| Organization-Centric Disadvantages | Trial-Centric Disadvantages |
| • Requires a formal registration process, as the high-level administrator needs to be validated by EMA and appointed in the EMA account management portal | • Less convenient when an organization applies for or runs multiple trials; possibility for duplication of data • Potential for impact on data quality and integrity by providing any user the option to create a clinical trial application with a sponsor organization if a sponsor administrator has not been registered and validated by the EMA |
There are several CTIS-related changes and challenges that, without knowledge of and advance preparation for, will significantly impact clinical trials.
Below are the main challenges with helpful tips for sponsors:
- Processes: The CTIS introduces tight response timelines. For example, responding to a request for information should be submitted through CTIS within a maximum of 12 days. If this timeline is not met, the application lapses. To avoid this and ensure appropriate coordination of responses, we recommend centralized access and communication via a dedicated and focused team. This team is then able to closely monitor timelines and the CTIS for incoming communications, handle document and data entry, and manage downloads for trial master file compliance.
- People: Sponsors will need to invest in training stakeholders on how to use CTIS and on change management processes.
- Trial Transitions: The transition of a trial from Directive 2001/20/EC to the EU CTR cannot happen during the assessment period of a substantial amendment, so the harmonization of study documents needs to be pre-planned.
- Technical Challenges: From the perspective of a sponsor or clinical research organization, there are various technical challenges of the CTIS, which will hopefully resolve with time. One example is the requirement for manual data entry; CTIS does not have an application programming interface, nor does it allow for upload of structured data (e.g., an XML file accompanied with submission package) that would populate data and document elements in the CTIS.
Another challenge is that the CTIS does not send alerts or notifications to users’ email addresses, forcing users to continuously check their studies for incoming alerts, requests for information and notifications. Considering the tight response timelines for some activities, it’s business critical to become aware of such study-related issues that require users’ action.
In addition, the EU CTR will significantly impact the submission preparation and tracking process. Technology needs to be adjusted for new types of submissions, new flows of document preparation, data approval, study data collection and more.
A few new study milestones have been introduced by the EU CTR, triggering submission of notifications to the regulatory authorities via the CTIS, so the sponsor must be able to capture these in a timely manner. Certain documents will no longer be received in a document or letter format (i.e., queries). so sponsors must find a new way to file them in an eTMF (electronic trial master file); perhaps only a different file format is needed (e.g., an image taken as a CTIS screenshot).
Although the CTIS rollout is a great milestone on the path to streamlining and improving clinical trial management and governance in the EU, we expect its technical capabilities to require several rounds of improvement to enhance process efficiency and allow more flexibility.
Taking into account the CTIS go-live date of Jan. 31, 2022, and the three-year transition period, we recommend that sponsors take early action by:
- Analyzing current business processes
- Adapting and upgrading their information systems
- Transforming operations to prepare for and avoid disruption to startup and maintenance of new and ongoing trials
Engaging with well-prepared partners can assist sponsors in navigating this exciting but challenging implementation.
We are here to help you navigate the changes brought about by the EU CTR.
We have additional resources you can explore, like this overview of CTIS opportunities and challenges and important changes and action items for sponsors. You can also explore how the EU CTR impacts investigational medicinal product labeling.